
Xavier RAFFOUX
dernières mises à jour 07/03/2023
xavier.raffoux@inrae.fr
Biologie de l'Adaptation et Systèmes en Évolution
Atelier Cartographie, Expression et Polymorphisme
INRAE, Ingénieur d'étude
publications - orcid
Thématiques:
- Etude du déterminisme génétique du nombre et de la position des évènements de recombinaison méiotique et des interactions entre recombinaison et évolution lors de l’adaptation.
- Analyse de données de type « Omics » pour l’unité : Bioinformatique et statistiques
- Conduite de projets de biologie moléculaire pour l’unité
Formation
- Doctorat en Sciences de la vie et de la santé, Université Paris-Sud (Orsay, France), 2018
- Master Biologie - Génome, Cellules, Développement, Évolution, Université Paris-Sud (Orsay, France), 2013
- Diplôme d’études universitaires générales en Biologie, UPMC(Paris, France), 1999-2000
Expériences
- Technicien de recherche, GQE-Le Moulon (50% ACEP, 50% RAMDAM)
- Etudiant en thèse, GQE-Le Moulon (Gif-sur-Yvette, France), 2013-2018
- Technicien de recherche, GQE-Le Moulon (Gif-sur-Yvette, France), 2003-2012
- Technicien animalier, LBMC Laboratoire de Biologie Moléculaire et Cellulaire, (Lyon, France), 2002-2003
Projet de recherche sur la Méiose
Une des thématiques de l’équipe a pour objectif l’étude du phénomène de recombinaison méiotique puisque c’est un des acteurs majeurs de l’évolution des génomes. En effet, pour que chaque gamète contienne un jeu complet de chromosomes, des liens physiques doivent être créés, lors de la méiose, entre les deux exemplaires (homologues) de chaque chromosome. Ces liens conduisent à un échange réciproque de matériel génétique entre chromosomes homologues que l’on appelle crossing-over. C’est ainsi qu’une partie de l’information génétique, originaire de deux individus parents, peut être combinée sur un même chromosome, et donc chez un descendant qui présente alors une nouvelle combinaison de caractères qui n’existait pas chez les parents. La reproduction sexuée permet ainsi la création de nouveaux assemblages de caractères chez les descendants, et cette nouvelle diversité est ensuite soumise à la sélection, qu’elle soit naturelle ou dirigée par l’homme, par exemple dans une optique agronomique. De plus, de nombreuses études génétiques exploitent les évènements de recombinaison afin d’identifier les régions du génome responsables de la variation de caractères d’intérêt, et la précision de ces études dépend en partie du niveau de recombinaison. Pour ces raisons, comprendre les facteurs génétiques contrôlant le nombre et la position des évènements de recombinaison est un enjeu crucial pour la médecine, l’agriculture, ou la recherche fondamentale. Une partie de l’équipe dont je fais partie étudie donc par des approches théoriques et expérimentales la formation des crossing-overs méiotiques, la régulation de leur nombre et de leur distribution, les relations entre recombinaison et évolution et enfin leurs implications pour l’amélioration des plantes.
Plus particulièrement, le projet ANR EvolRec qui a commencé en 2021, en rapport avec cette thématique pose les questions scientifiques suivantes chez la levure S. cerevisiae :
- Comment le taux de recombinaison répond-il à une sélection directionnelle ?
- Quels sont les déterminants génétiques du nombre et de la répartition des crossing-overs ?
- Quelles sont les interactions entre recombinaison et évolution lors de l’adaptation ?
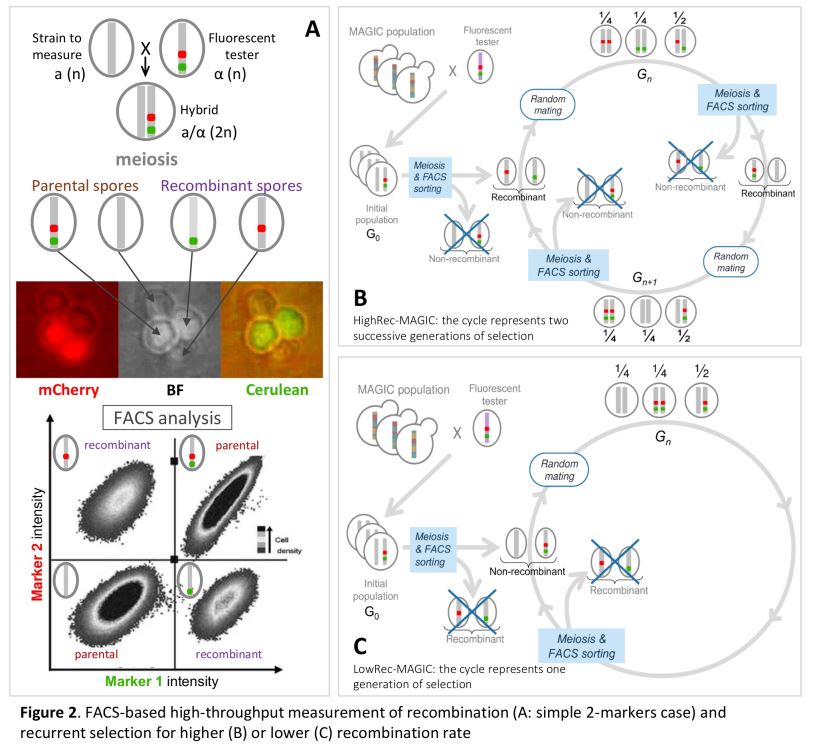
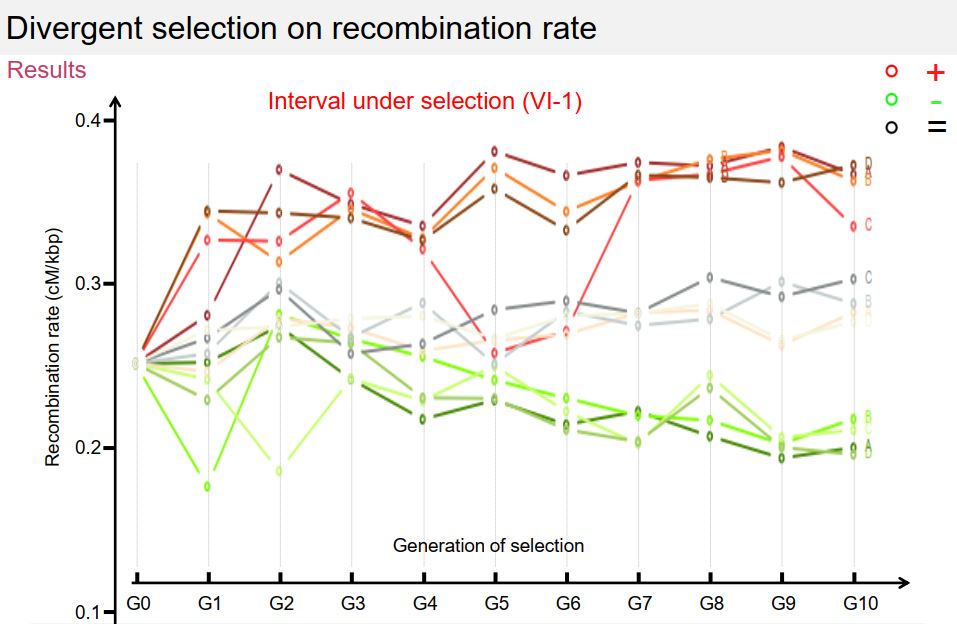
Dans le cadre de ce projet, je poursuis les travaux initiés pendant ma thèse sur le contrôle du nombre et de la position des crossing-overs et je développe de nouvelles approches visant à étudier les relations entre la recombinaison et l’adaptation via l’évolution des génomes. Ainsi, je conduis depuis plusieurs années une expérience de sélection récurrente sur le taux de recombinaison chez S. cerevisiae afin (1) d’étudier la réponse à la sélection de ce caractère, et (2) de détecter des QTLs de recombinaison (Quantitative Trait Locus = région du génome associée à la variation d’un caractère quantitatif). Dans ce cadre, j’ai mis au point une méthode originale basée sur la cytométrie en flux, permettant de sélectionner à haut débit des spores selon leur statut recombinant ou non entre deux marqueurs fluorescents (300 000 individus triés en 15 minutes), puis de les inter-croiser en panmixie pour former la génération suivante. Ainsi, il est maintenant possible de sélectionner de manière récurrente des spores selon leur statut de recombinaison, ce qui lève un verrou technique et ouvre une nouvelle voie pour l’étude du déterminisme génétique du taux de recombinaison. Grâce à cette approche, j’ai réalisé 11 générations de sélection récurrente selon trois modalités : (A) sélection des individus ayant recombiné entre les marqueurs, (B) sélection des descendants n’ayant pas recombiné entre les marqueurs et (C) sélection des descendants indépendamment du niveau de recombinaison entre les marqueurs. Les résultats obtenus pour les 12 populations et sur les 11 générations de sélection montrent que le niveau de recombinaison varie en réponse à la sélection dans la région chromosomique où la sélection est appliquée, et qu’il varie dans le sens opposé, et dans une moindre mesure, dans la région adjacente, ce qui suggère des effets locaux de compensation (interférence). En revanche, le niveau de recombinaison ne montre pas de variation à grande distance de l’intervalle sous sélection, ni sur les autres chromosomes étudiés. Enfin, cette variation n’est pas apparue progressivement au cours des générations, mais par paliers, et la recombinaison est revenue à son niveau initial en une génération lorsque la pression de sélection a été levée. Ces 13 populations ont été séquencées à haute couverture (50X) ce qui va permettre de détecter des différences de fréquences alléliques entre la population initiale et les populations évoluées d’une part, et entre les 3 modes de sélection d’autre part.
Toujours dans une thématique de recherche centrée sur la recombinaison méiotique, je participe à la réflexion sur un work package d’un second projet ANR (CO-PATT), également financé à partir de 2021, porté par Éric Espagne (I2BC, Orsay). Dans le cadre de ce projet, nous allons réaliser un crible suppresseur chez la levure pour détecter les gènes restaurateurs de l’interférence chez des mutants du gène slx5 qui réduit l’interférence entre crossing-overs.
Projets de biologie moléculaire
Estimation des fréquences de différentes variétés de blé, après semis puis récolte en mélange
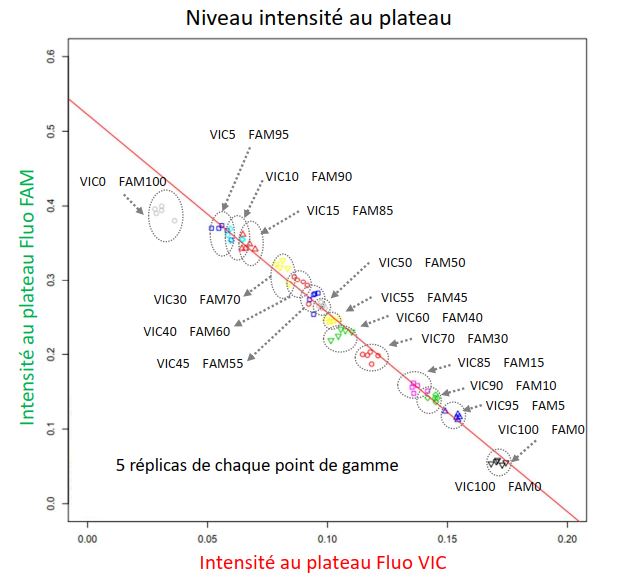
Premièrement, dans le cadre de la thématique de conception participative de mélanges variétaux de l’équipe Diversité, Evolution et Adaptation des Populations (DEAP), mon objectif est d’estimer la fréquence de différentes variétés de blé, après semis puis récolte en mélange. Nous avons choisi de concevoir une méthode combinant deux techniques existantes de biologie moléculaire : la PCR quantitative en temps réel et la PCR allèle-spécifique compétitive (KASP). J’ai développé un script R permettant de modéliser les données produites afin d’extraire certains paramètres tels que l’efficacité de la réaction, le niveau de fluorescence au plateau ou le Ct, ceci à partir des données brutes de fluorescence au cours des cycles d’amplification. Mon travail permet ainsi d’exploiter les données et d’estimer les fréquences de chaque variété à partir de deux des proxys extraits de la courbe d’amplification en temps réel (niveau de fluorescence au plateau et Ct). Les résultats, réalisées sur une gamme de mélanges binaires d’ADN de feuille de blé en proportions variables, montrent que nous sommes capables d’estimer ces fréquences avec 5% d’incertitude. Les objectifs fixés sont d’améliorer expérimentalement la précision de l’estimation puis de valider un panel de marqueurs moléculaires utilisables à plus haut débit.
A court terme, ce travail permettra de connaître les proportions de génotypes de blé présents en mélange dans la parcelle à partir d’échantillons d’ADN issus de mélanges de farine. Il sera alors possible d’étudier l’évolution des proportions de différents génotypes en mélange au cours des générations. C’est le cas des dispositifs expérimentaux où l’on utilise la récolte pour semer l’année suivante. Enfin, dans le cas des conduites agricoles où un nouveau mélange de grains est préparé chaque année, ce travail permettra d’étudier quels facteurs (intrants, parasites, stress abiotiques) peuvent influer la culture en compétition de plusieurs génotypes.
Vérification de génotype avant séquençage de novo
Deuxièmement, dans le cadre d’un projet visant à étudier comment les variations structurales du génome (éléments transposables, duplications de gènes) peuvent moduler le transcriptome, j’ai génotypé par microsatellites des lignées de maïs pour vérification de leur identité avant leur séquençage de novo. Pour ce projet, j’ai conduit en les adaptant les protocoles techniques existants, assuré la fiabilité des résultats (système de double lecture), mis en forme puis transmis les résultats aux chercheurs impliqués lors d’une réunion de fin de projet.
Développement de marqueurs microsatéllites chez le puceron cendré du pommier
Enfin, troisièmement, dans le cadre d’un projet visant à étudier la coévolution adaptative entre un parasite (puceron cendré) et un hôte soumis à domestication (pommier), mon objectif était de pouvoir identifier différents génotypes de pucerons et de vérifier leur mode de reproduction en chambre de culture. Pour mener à bien ce projet, en concertation avec le responsable de l’équipe ACEP, M. Falque, nous avons fait le choix de la technique, des marqueurs à utiliser et de leur nombre (dimensionnement de l’expérience en fonction des questions posées), ceci lors d’une réunion de début de projet avec le doctorant impliqué. J’ai alors produit les premières données (génotypage de pucerons cendrés sur 8 microsatellites), analysé et mis en forme les résultats.
Bioinformatique et analyses statistiques de données omics
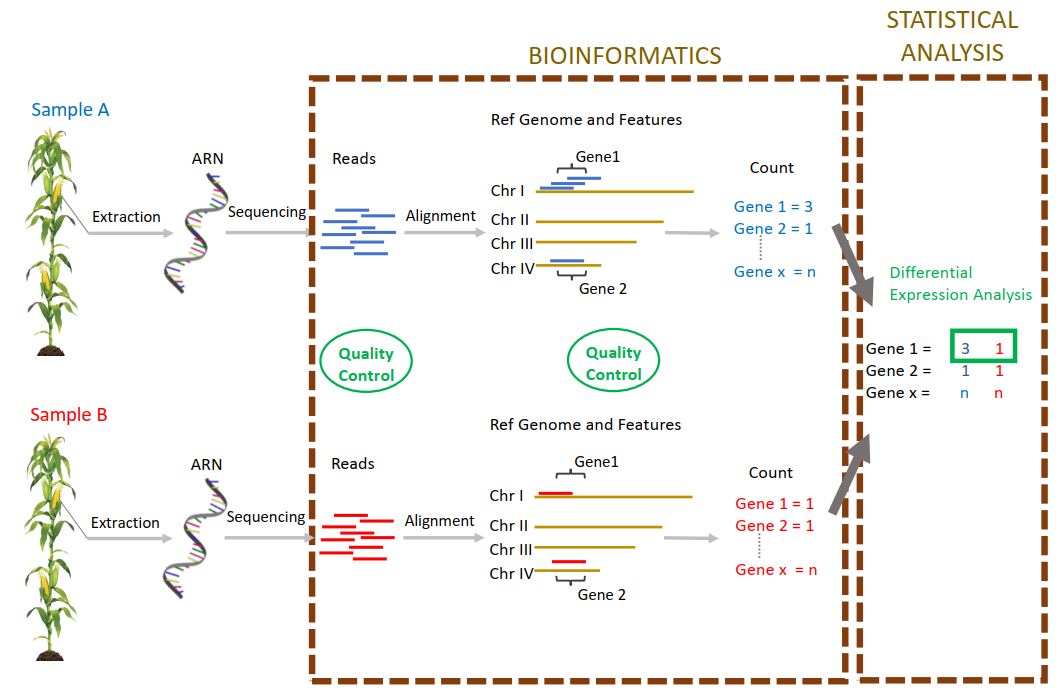
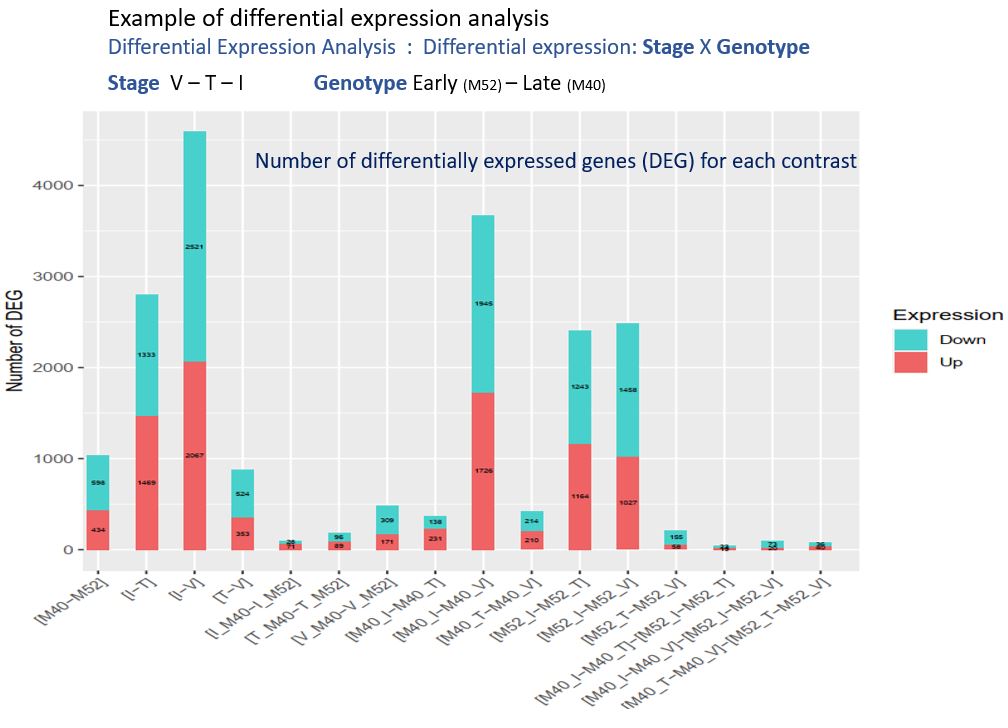
Actuellement, dans la cadre d’une des tâches du projet « Itemaize » qui vise à étudier les caractères cibles de la sélection pour la date de floraison chez le maïs, j’analyse des données de RNAseq provenant de deux génotypes de maïs. Ces deux génotypes sont issus d’une expérience de sélection divergente sur la précocité de floraison, expérience conduite depuis plus de 20 ans dans l’unité. Chaque année, des plantes sont ainsi sélectionnées sur la base de leur date de floraison, pour former la génération suivante. Mon objectif est d’étudier le niveau d’expression génique dans les méristèmes de plantes de ces deux génotypes (précoce – tardif), prélevés à la génération 18 et à différents stades de développement. Ainsi, il est possible de détecter les gènes différentiellement exprimés entre génotype précoce et tardif, entre stade de développement, et d’étudier les interactions entre ces deux facteurs. Les gènes détectés sont potentiellement impliqués dans la transition florale, caractère d’intérêt agronomique majeur dans le contexte de changement climatique. Plus en aval, l’étude de la réponse à la sélection au cours de cette expérience et des différents parcours évolutifs suivis nous renseignera sur les processus évolutifs mis en jeu dans le cadre de l’adaptation à un nouvel environnement.
Après un contrôle qualité des données ( FastQC et MultiQC), afin de détecter d’éventuels problèmes de séquençage ou biais expérimentaux, j’ai aligné les reads sur le génome de référence.
Pour cela, j’ai choisi un logiciel d’alignement adapté (STAR), conçu pour effectuer cette tâche sur des génomes complexes tels que celui du maïs.
Celui-ci est en effet un génome eucaryote d’une taille de 5 000 Mpb, avec la présence de gènes dupliqués et d’éléments transposables.
De plus, ce logiciel utilise les informations d’annotation du génome pour effectuer l’alignement, mais peut aussi découvrir de nouveaux introns non encore annotés.
Ces données m’ont permis de produire un tableau de comptage (nombre de reads par gène et par échantillon) en utilisant le logiciel « HtSeqCount » et les paramètres permettant un comptage qui soit le reflet le plus juste du niveau d’expression des gènes.
Des analyses en composante principales sur les données brutes de comptage m’ont permis d’observer que les données sont structurées selon nos deux facteurs (Génotype et stade de floraison) mais pas entre les réplicas biologiques, ce qui fait sens dans cette expérience. Un des échantillons a été écarté des analyses suivantes car trop différent des autres.
J’ai ensuite écrit un script R utilisant la librairie DiCoExpress pour analyser statistiquement les données de comptage et ainsi détecter les gènes montrant une expression différentielle dans le contexte de la transition florale. Pour cela, j’ai normalisé les données brutes de comptage (méthode TMM), définit un modèle, et un seuil de détection. Pour chacun des contrastes étudiés, j’ai observé la distribution des p-values et vérifié ainsi que le seuil de nombre de reads minimum par gène était pertinent. J’ai ensuite recherché dans la bibliographie des informations sur la fonction de certains des gènes détectés et trouvé des informations en rapport avec la transition florale. Ce travail ne pouvant pas être réalisé pour l’ensemble des gènes détectés, j’ai fait un travail d’ontologie en replaçant chaque gène dans son groupe de fonction. Ce travail permet d’avoir une vision d’ensemble et de comprendre quelles voies métaboliques sont régulées différemment au moment de la transition florale.
Je vais prochainement choisir puis utiliser les analyses statistiques permettant de continuer mon travail à une échelle multi-omiques, avec par exemple l’intégration des données de phénotypage.
Les résultats obtenus seront ensuite couplés à des données métabolomiques et protéomiques afin de construire un réseau de régulation de la transition florale.
Publications
- Olvera-Vazquez SG., Alhmedi A., Miñarro M., Shykoff JA., Marchadier E., Rousselet A., Remoué C., Gardet R., Degrave A., Robert P., Chen X., Porchier J., Giraud T., Vander-Mijnsbrugee K., Raffoux X. , Falque M., Alins G., Didelot F., Beliën T., Dapena E., Lemarquand A., Cornille A.. (2021) Experimental test for local adaptation of the rosy apple aphid (Dysaphis plantaginea) to its host (Malus domestica) and to its climate in Europe. PCI Ecology, (Pre-registration version)
- Raffoux X. , 2018-06-11 06/11/18, Diversité et déterminisme génétique de la recombinaison méiotique chez Saccharomyces cerevisiae, PhD thesis, Université Paris-Saclay
- Raffoux X. , Bourge M., Dumas F., Martin OC., Falque M.. (2018) High-throughput measurement of recombination rates and genetic interference in Saccharomyces cerevisiae. Yeast, 6 (35) 431-442
- Raffoux X. , Bourge M., Dumas F., Martin OC., Falque M.. (2018) Role of Cis, Trans, and Inbreeding Effects on Meiotic Recombination in Saccharomyces cerevisiae. Genetics, 4 (210) 1213-1226